Overview
Asthma is a chronic inflammatory disease characterised by bronchial hypersensitivity, bronchspasm and mucous overproduction
It is a complex, multifactoral disease with genetic and environmental factors. It is also a spectrum of disease with there being multiple different subtypes and severity within a subtype. Common symptoms are asthma attacks and airway obstruction.
Types of Asthma
There are many different types of asthma:
Work related asthma
As the name states this is asthma which is related to work. This can further be divided into Occupational asthma, or work exacerbated asthma. Occupational asthma can be sensitizer induced or irritant induced, and work exacerbated asthma is just asthma which triggered at work.
T2 type vs non T2 type asthma
T2 type and non T2 type asthma refers to the
Pathogenesis
So this pathogenesis is rather complex I think, but I will do my best to detail it (This is in incredible oversimplification and is full of nuance)
- Dendritic cell presents antigen to naive T helper cell (Th-0 )
- It can either differentiate into Th-1 or Th-2 cells.
Th-1 response (IFN-γ, lymphotoxin, IL-2)
This is cell-mediated immunity and leads to neutrophilic inflammation. It is selected for with IL-12 which also inhibits the Th-2 response.
Th-2 Response
This is the other one which very complex but leads to asthma symptoms. Triggers an antiparasite type reaction,
This response provokes synthesis and release of proinflammatory cytokines to prime B lymphocyte cells, drives maturation of dendritic cells, increases numbers of inflammatory cells recruited and increases IgE levels which worsens asthma symptoms.
The released cytokines also provoke autonomic system to bronchoconstriction.
Asthma priming?
After the initial Th2 response the airways can become sensitised. This happens via B cells making IgE antibodies to an allergen. which is then attached to mast cell, eosinophils and basophils. Inflammatory response occurs upon second contact.
Clinical profile:
Clinical Signs and Symptoms:
Pathological profile:
Pathophysiology
Airway inflammation Bronchial hyper resposible bronchocontriciton Bronchial wall edema excess mucous secretion epithelial shedding → bronchospasm airway remodelling → more goblet cells matrix and smooth muscle\
Exacerbations
Starts with Type I hypersensitivity with ige triggered mast cells
Long term develops into type IV hypersensitivity
Genetic profile
Public Health profile
Treatment of asthma
finish need to sort the chemical basis of asthma out to own section and then clean up
Asthma is an inflammatory disease with bronchconstriction and overproduction of mucous, and can result in remodelling.
To understand how to treat asthma you must look how at the mechanisms by which it works and perpetuates itself.
The Th-2 response leads mast cells basophils and eosinophils to release mediators of inflammation and this will happen in the future due to desensitisation. These are histamine, prostaglandins, leukotrienes and enzymes. Just think about asthma reaction = inflammatory mediators at the moment.
Overview
- Primarily manage the inflammation (use inhaled corticosteroids (ICS))
- Secondarily reserve bronchodilators for symptomatic relief (prodilators like LABAs and SABAs, anti-constrictors like SAMAs LAMAs)
Proinflammatory cytokines
Both prostaglandins and leukotrienes are both made of arachidonic acid which in turn is made from membrane phospholipids
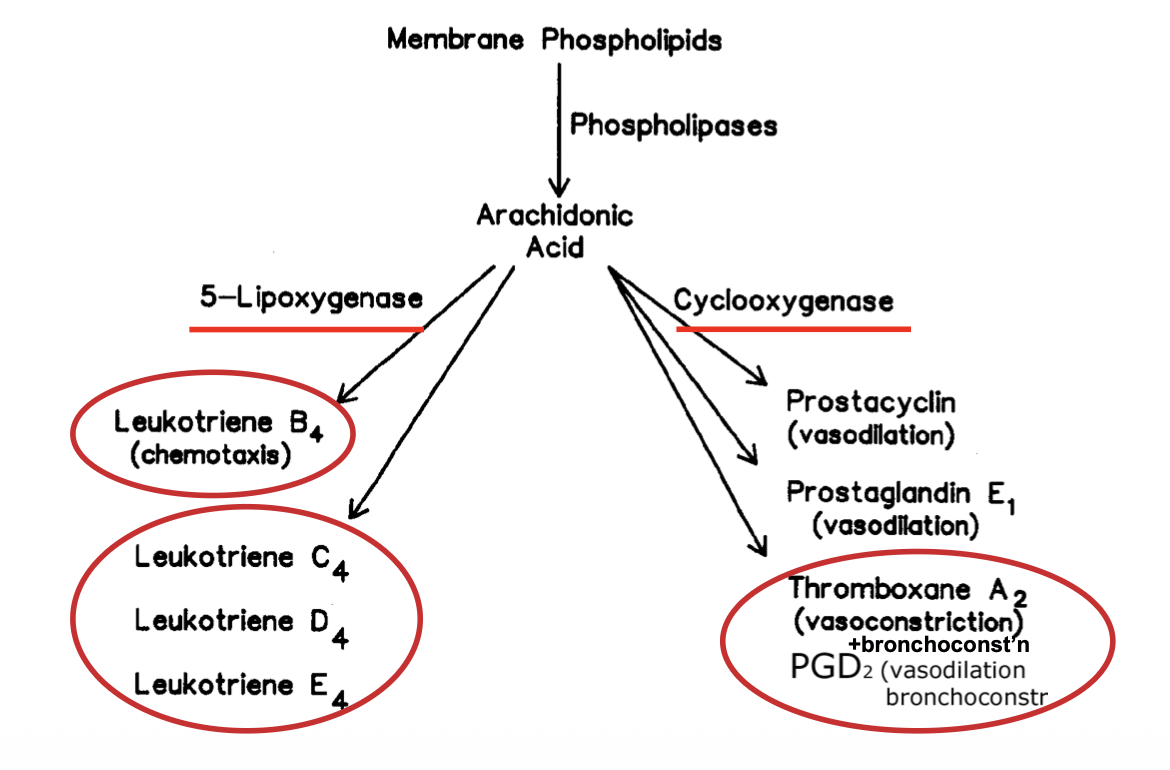
Leukotrienes
These are “slow reacting substance of anaphylaxis” and cause contraction of respiratory smooth muscle, and increased vascular permability.
Prostanoids (prostaglandins+thromboxanes)
PGE1
PGE1 promotes vasodilation and also down-regulates the lipoxygenase pathway, resulting in less leukotrienes
Drug-induced rhinitis
As NSAIDs inhibit COX enzymes this can lead to an increase in leukotrienes, which can result in “Non-Allergic Rhinitis with Eosinophilia syndrome”. This is charaterised by rhititis( nasal obstruction rhinorrhoea, sneezing) and facial flushing.
Ensuing asthma triggered 1-3 hours within ingestion of aspirin adn other NSAUDS
Prostaglandins (PGD2)
These mediate bronchoconstriction and vasodilation, they increases permeability and therefore inflammatory cells infiltration.
Thromboxanes (TXA2)
These mediate bronchoconstriction
Bronchial tone regulation
Bronchial smooth muscle can either constrict or dilate
Dilation
In Asthma attacks this isnt happening and we want it to happen. cAMP leads to Bronchodilation. Its production is upregulated by b adrenoreceptor activation which we can stimulate with ß2 Adrenoreceptor agonists. (LABA, long acting ß2 agonist, SABA, short acting ß2 agonist)
Constriction
This is upregulated by adenosine and acetylcholine (the big one). We can limit this with muscarinic antagonists, which block neural bronchocontriction. (LAMAs, Long acting muscarinic antagonist, SAMAs, short acting muscarinic antagonist)