Derived from Blood Lecture 6, This lecture is dually an introduction to haemostasis and to platelets. It also introduces coagulation, thrombocytopenia (low platelets), and platelet functional abnormalities. it mainly talks about platelets though.
Haemostasis
What is Haemostasis?
Haemostasis is complex process which results a leak from a blood vessel being stopped. The word etymology is haemo- (blood) -stasis (stopping) and so means blood stopping. Its purpose is to stop active leaks in blood vessels but can often be pathological, as thrombi (Blood clots inside your vessels) can form and turn into embolise (break off and cause and embolism somewhere like a stroke).
Mechanics of haemostasis
Haemostasis has 3 main components: 1. vessel constriction, 2. Primary haemostasis (platelet activation and plug), and 3. secondary haemostasis (coagulation cascade). These are introduced more in depth below. (However most of the rest of this lecture is primary haemostasis and the next is secondary haemostasis)
1. Vessel constriction
As we have seen in the physiology lectures, the main determining factor of vessel resistance to a traveling substance (liquid or gas) is radius. In haemostasis you want the blood to flow less to the area, to curb active bleeding and to allow platelets to marginate.
2. Primary haemostasis
Primary haemostasis is the activation and aggregation of platelets forming a plug to immediately “plug” a hole in the blood vessel. These activated platelets also play a role in the activation of the next step, coagulation. Additionally, too much platelet activation can lead to arterial thrombosis and too little can lead to bleeding from mucosal surfaces, gut bleeds, bruising, and menorrhagia. Speaking of, platelets in very important in stopping bleeding from mucosal surfaces. This is the first step in healing
3. Secondary haemostasis
This is what is referred to as coagulation, is a complex process which results in fibrinogen getting catalysed into fibrin and deposited on the platelet plug. Coagulation is NOT the same as haemostasis, it is a part of haemostasis. Additionally it does not directly involve platelet, but they are critical to this. Coagulation relies on a protein cascade (Coagulation cascade) which results in an enzyme called thrombin which cleaves fibrinogen into fibrin, which makes a gel. Too much coagulation cascade can lead to thrombus forming in veins (eg deep vein thrombosis) and too little can lead to bleeding disorder eg haemophilia.
Platelets (primary haemostasis)
Initation of primary haemostasis
Usually the endothelium of an artery (or any blood vessel? recheck ) is like teflon, it is nonstick. But when there is damage to the endothelium a number of things happen to trigger platelet activation:
- NO and PGI2 stop being released. these are potent antiplatelets and vasodilators which are released in a normal state/
- ADP and TxA2 are release which are proplatelet activation (I think they are pro margination recheck) These 2 things (there is probably more at play) really get the next step going which is platelet binding to ECM collagen through Von willebrand Factor (vWF). This is called platelet activation. vWF is like a connector which allows platelets to bind to collagen.
N.B. Platelets can bind directly to collagen but it is slower and less strong. Lowkey Oyelese didn’t initially mention this and it took me a little while for this to click.
Platelet activation
Platelets can activate in a number of ways. They can activate through binding to collagen, other platelets (for aggregation) and fibrin (also for aggregation).
After platelet activation, the platelet changes into the dendritic form and releases 2 types of granules containing a number of molecules listed below:
Platelet granule release
Alpha granules
- vWF
- fibrinogen and factor v
- Platelet derived growth factor - to recruit fibroblasts for healing
- factor 4 - healing
- transforming growth factor b - healing Dense or Delta granules
- ADP
- serotonin
(This is a non-exhaustive list)
These molecules lead to the activation of more platelets, as well as starting the coagulation cascade.
Dendritic form
As previously mentioned the platelet morphs into its dendritic form, which allows for more contact surface areas for crosslinking with other platelets.
Aggregation
The activated platelets will continue to attract and bind to other platelets, eventually forming a plug which is cemented in place by fibrin from the coagulation cascade (which was in part activated by activated platelets), which is eventually remodelled into vessel wall once again.
To summarise
Injury
- Endothelium is damaged
- Prostacyclin and NO stop being released
- ADP and TxA2 are released
Initiation
- Platelet recruitment
- vWF attaches the platelet to the collagen via platelet receptor
- This causes conformational change in the platelet from resting to dendritic
- t\The dendritic platelet then releases molecules causing bonding and activation of and with other platelets
Extension
- Recursive effects of more binding and coagulation cascade
- ADP (Causes crosslinking between platelets) and TxA2 (thromboxane A2, a vasoconstrictor and platelet activator)
- Coagulation cascade thrombin released as part of this
Stabilisation
- fibrinogen is triggered by thrombin to form fibrin which cements the plug together ans in place
Other notes…
Key Platelet receptors
These are key receptors on the platelets relating to and reinstating whats been said before. I think they all lead to activation?
- vWF Receptor: for initial adhesion and activation
- Collagen receptor: for initial adhesion and activation
- ADP receptor: secreted by adjacent platelets
- Thrombin receptor: a product of coagulation
- Thromboxane receptor: secreted by adjacent platelets
- Fibrinogen receptor: formation of bridges between platelet (I think platelet use fibrinogen as a bridge to help with platelet to platelet connection? recheck)
Steps of platelet activation
These are the intracellular (can I say that about platelets?) steps of platelet activation ( dont particularly understand this):
- Adhesion to vWF, or somthing similar
- increase in intracellular calcium
- increased phosphatidylserine exposure to create a negatively charged surface for coagulation
- cytoskeletal rearrangement
- alpha and delta granule release
- thromboxane a2 generation
Platelet production 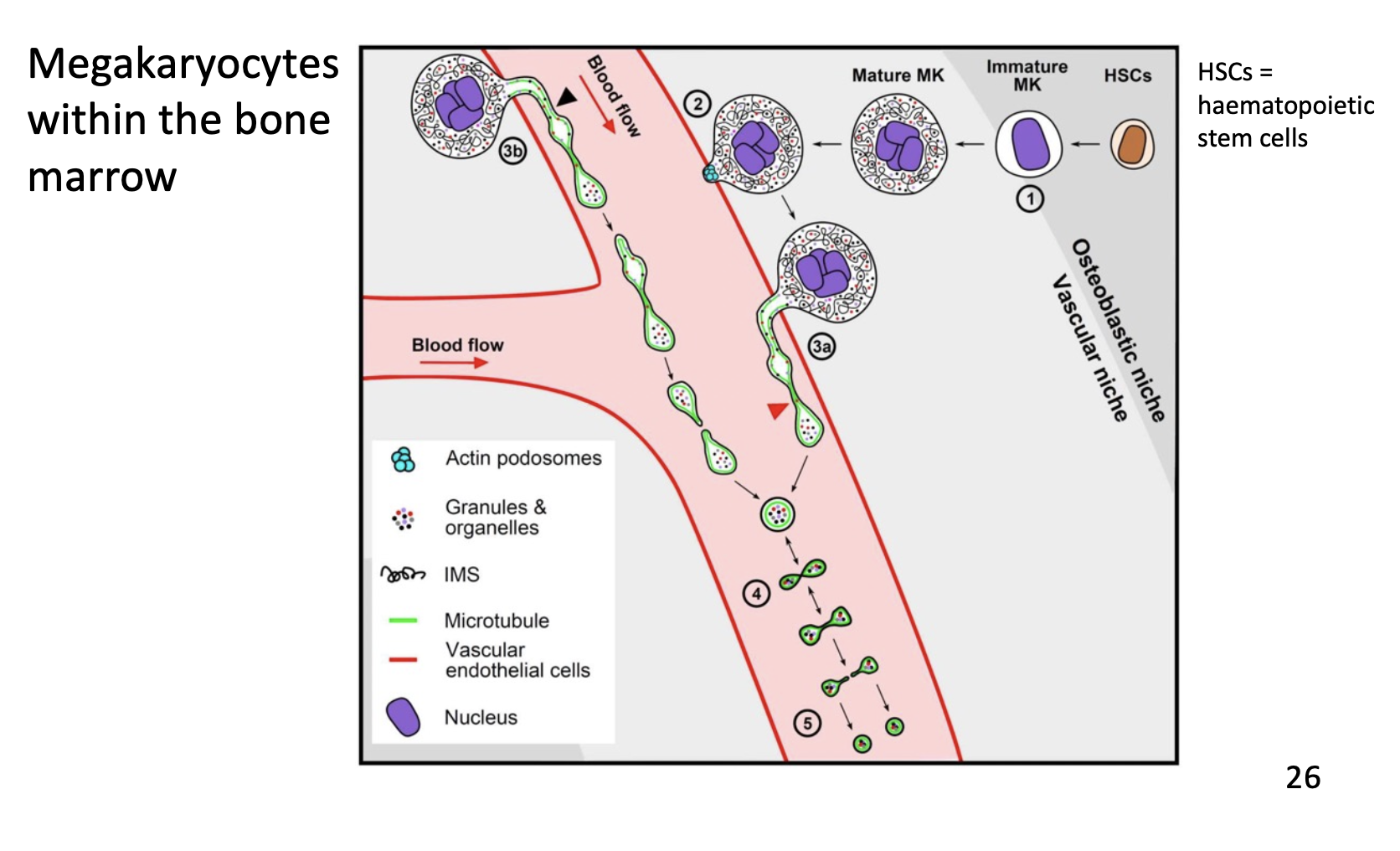
Platelets are produced from megakaryocytes. These cells “bud” their cytosol and plasma membrane off into the blood vessels almost akin to blowing bubbles into the breeze, which form proplatelets. These mature into full platelets. This process can be stimulated by thrombopoietin (TPO)
Megakaryocytes have massive nuclei and reside in the bone marrow.
Thrombocytopenia
Thrombocytopenia is when there are low platelets in the blood/ body.
Manifestations
They can lead to petechiae, purpura, and ecchymoses.
- Petechiae are small pinpoint bleeds resulting from microtears in vasculature which cannot be plugged as there is nothing to plug them. A disseminated intravascular coagulation can lead to petechiae, especially when its sepsis induced.
- Purpura are the medical term for bruising
- Ecchymoses is a large patch of purpura
Causes of low platelets
The causes of reduced marrow production are as follows, reduced marrow production (see next bit), immune destruction (this can be from immune thrombocytopenia, or drug associated immune destruction), DIC (see DIC page), genetic, and splenomegaly (normally the spleen holds 25% of platelets but this can rise to 90% in the event of splenomegaly)
Reduced production
Reduced production of platelets can be due to: marrow depression, which can be caused by chemotherapy, radiotherapy, drugs and chemicals etc., marrow failure, due to marrow replacement (eg cancers invading and pushing out other cells), and selective depression of megakaryocytes (eg. drugs and chemicals, and some viral infections like chickenpox, or Epstein-Barr virus)
Typical levels for platelets
| Level | Concern or not |
|---|---|
| 150-400 | Normal |
| 100-150 | No worries |
| 50-100 | Caution with surgery or trauma |
| <50 | Bruising is common and all anticoagulants or antiplatelets should be ceased |
| 10-20 | Extra caution, Platelet transfusion may be needed |
| <10 | Platelet transfusion now. |
Platelet functional abnormalities
Platelets can be found in normal quantities, yet can still result in poor clotting. This will be inherited or acquired.
Inherited
The main inheritable platelet dysfunction is Von Willebrand disease. It is autosomal dominant and results in low levels of vWF. It effects 1 in 500 to 1 in 1000 people.
There are a number of other platelet defects all which can cause platelet deficiency.
Acquired
Acquired platelet dysfunction is usually the result of drugs or foods.
Aspirin (cyclooxygenase inhibitor) and clopidogrel (ADP receptor blocker) are both examples of anti-platelet drugs.
NSAIDS in general reduce platelet function
PFA (Platelet Function Analyser) test
Platelet function can be measured via this in-vitro test. It involves a capillary tube coated with collagen-adrenalin or collagen-ADP and fresh citrated blood is drawn though this capillary tube. The time it takes for a clot to occur is a measure of how well the platelets work. Collagen-adrenaline should take 82-160s and collagen-ADP should take 62-120s.